Axl S. Cepeda

In January 2021, I started my Ph.D. under the guidance of Dr. Ananias A. Escalante at Temple University. Research at the Escalante's Lab focuses on the ecology, epidemiology and evolution of infectious diseases. I am interested in establishing bridges among population genetics, ecological, and evolutionary biology perspectives by interpreting genomic patterns from pathogens (mainly in Plasmodium spp.) and integrating them with environmental or epidemiologic information.
Escalante-Pacheco Lab 👨🏼🔬👨🏽💻
Department of Biology, iGEM 📊👩🏻⚕️
Temple University 🏫🇺🇸
U. Nacional de Colombia, GERPH 🎓🇨🇴
View My Google Scholar Profile
My Research at Temple University
Applying a Novel Sequencing Pipeline to Uncover Parasite Diversity in Owls
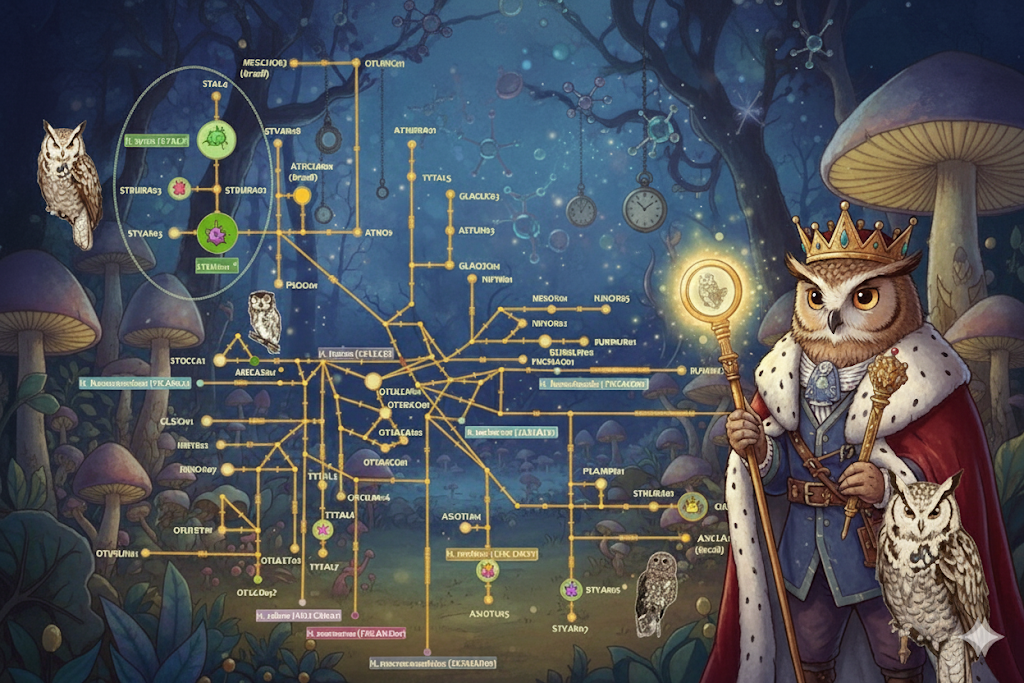
Building on our development of a high-throughput sequencing pipeline, this study applied our new method to a real-world biological problem: uncovering the hidden diversity of malaria-like parasites in North American owls. By analyzing the complete mitochondrial genomes using our PacBio HiFi protocol, we successfully detected complex mixed infections and revealed a rich diversity of nine distinct parasite lineages, including two new to science. A key discovery was linking the common genetic lineage hSTVAR01 to the morphospecies Haemoproteus syrnii for the first time. Our data also revealed that what was previously thought to be a single species is actually a complex of multiple cryptic species, demonstrating the power of high-quality genomic data to resolve hidden biodiversity. [Publication]
A Machine Learning Pipeline for High-Throughput Sequencing of Malaria Parasites
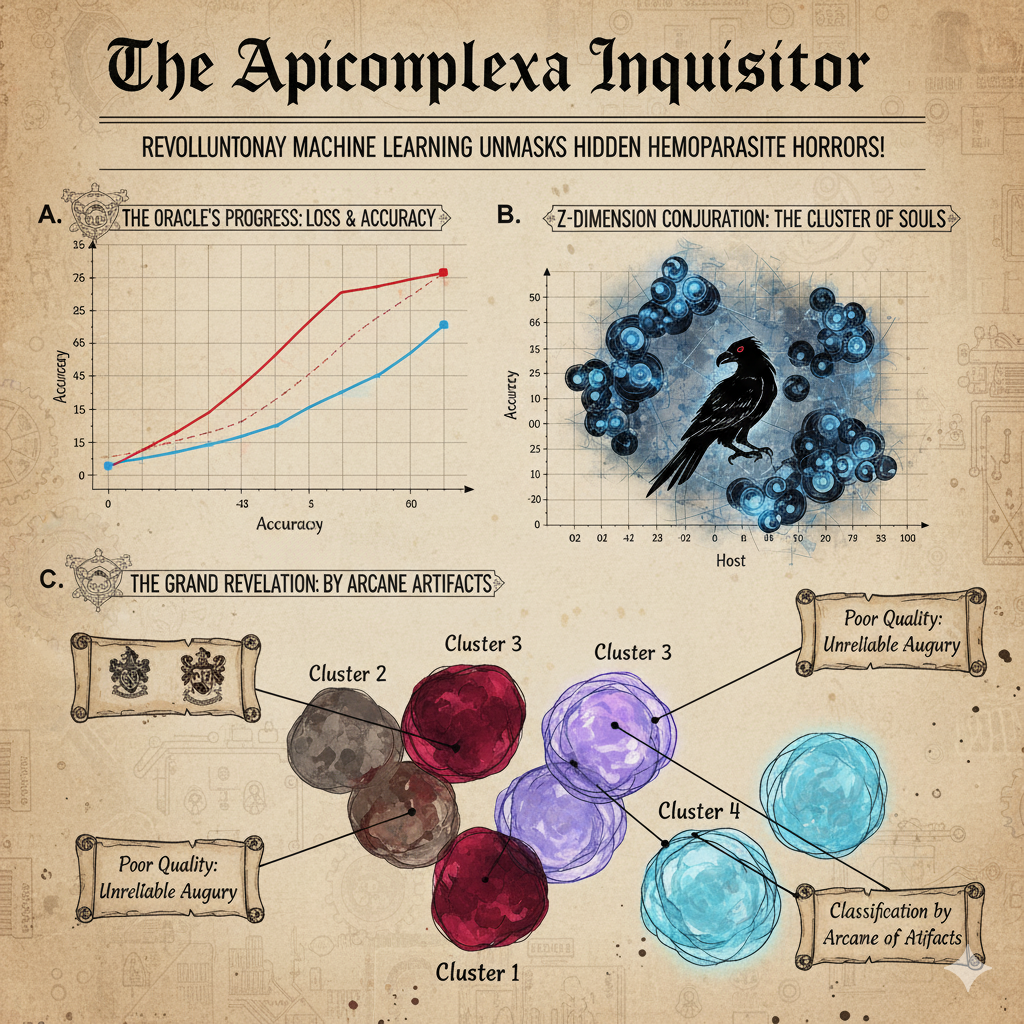
Traditional DNA barcoding of malaria-like parasites often fails to detect complex co-infections and mixed-strain infections, limiting our understanding of their true biodiversity. To address this, we developed a new high-throughput sequencing protocol targeting the entire mitochondrial genome with PacBio HiFi long-reads. My primary contribution was the design and implementation of the HmtG-PacBio Pipeline, a novel bioinformatics workflow at the core of this method. This pipeline utilizes a machine learning model (a variational autoencoder) to process and automatically classify the long-read sequences, accurately sorting reads to identify distinct parasite haplotypes and species within a single complex sample. The method successfully characterized infections from diverse hosts—including birds, reptiles, and mammals—that were missed by standard techniques, providing a powerful and scalable new tool for biodiversity and population studies of haemosporidian parasites. [Publication]
The Genome of an African Monkey Malaria Parasite Rewrites Evolutionary Timelines
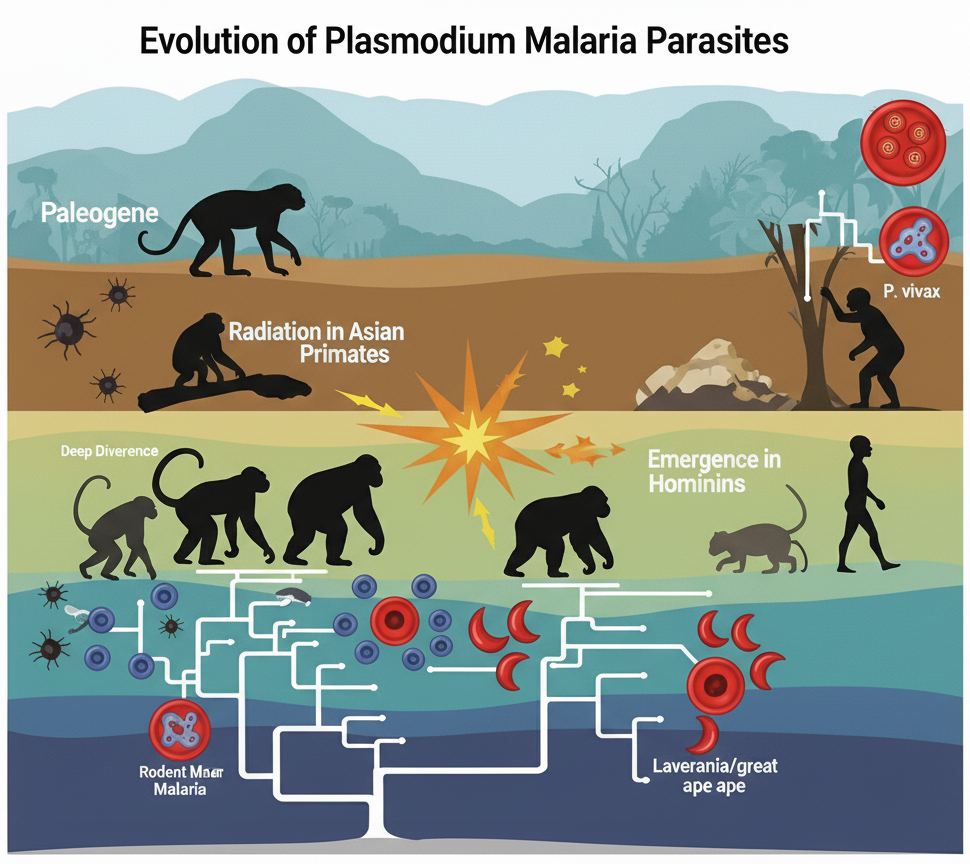
As a central part of my doctoral research, I generated the first chromosome-level de novo genome assembly for Plasmodium gonderi, a key malaria parasite of African monkeys. This high-quality genome served as a critical new resource to investigate the evolutionary origins of human malaria. My phylogenomic analyses placed the common ancestor of the human parasite P. vivax within the radiation of Asian primate malarias, strongly supporting an Asian origin for this widespread species. Furthermore, my time-tree analyses provided new estimates for major evolutionary events, dating the origin of the lethal P. falciparum clade to the same period as the radiation of its great ape hosts. [Publication]
A Tale of Two Clades: The Deep Evolutionary Divergence of Human Malarias
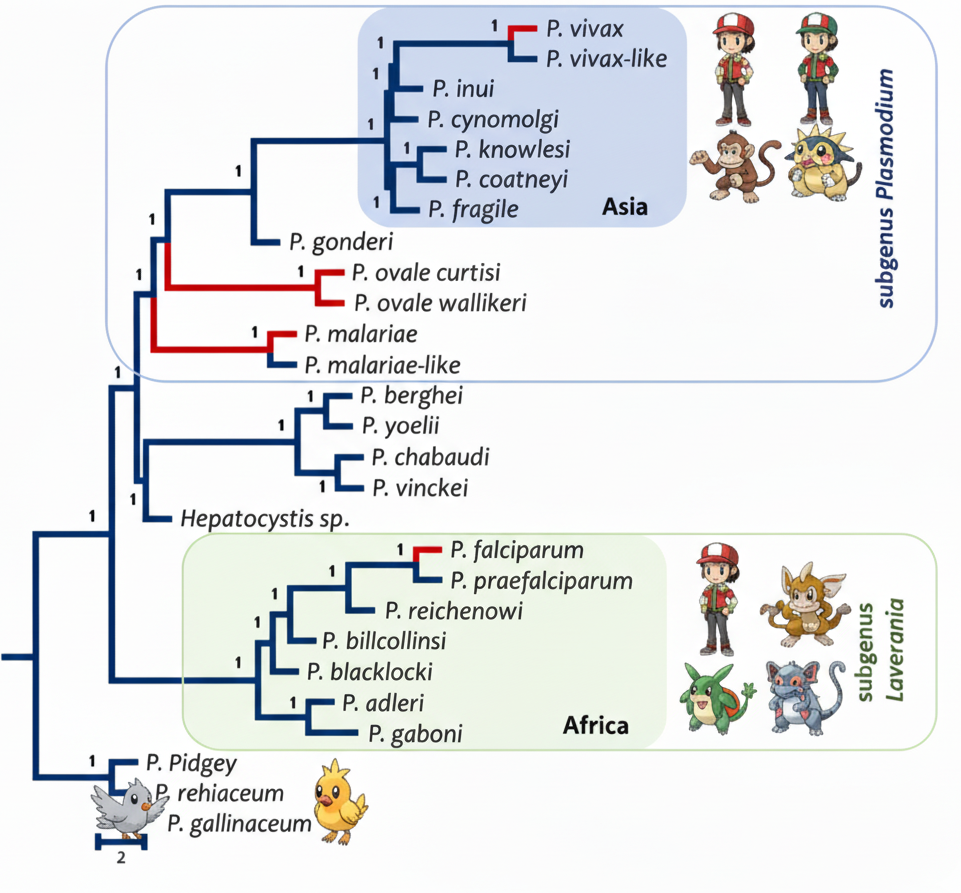
This review explains why the two main human malaria parasites, P. vivax and P. falciparum, are so biologically different. We show their distinct traits stem from a deep evolutionary split into two clades: subgenus Plasmodium and Laverania. My contribution was performing the phylogenomic reconstruction of the species tree from over 1,028 genes. This analysis provided the robust framework to visualize their ancient divergence and contextualize the unique molecular adaptations in each lineage. [Publication]
Uncovering Hidden Parasite Diversity in Turtles Through Genomics
Standard genetic markers are often insufficient to identify species of Haemogregarina blood parasites in turtles. To solve this, our project developed novel mitochondrial markers. I executed the core bioinformatic pipeline, performing the de novo assembly of the parasite's mitochondrial genome from raw sequencing reads after filtering out host data. This assembled genome served as the essential blueprint to design new primers, which successfully uncovered a vast, previously hidden genetic diversity. [Publication]

